一种以荧光激活模式运行的基于 Cas6 的 RNA 跟踪平台
目前活细胞RNA追踪技术分为两种:荧光富集(FE)型和荧光激活(FA)型。FE 型平台通常包含两个关键组件:充当报告器或跟踪器的荧光蛋白偶联 RNA 结合蛋白 (RBP-FP),以及在目标 RNA 上遗传构建的特定 RBP 结合序列 (RBS)。FE 型的一个突出例子是 MS2-MCP RNA 跟踪系统,其中 MCP 代表 MS2 噬菌体外壳蛋白(1)。MCP 结合称为 MBS(MCP 结合位点)的特定 RNA 茎环结构。为了跟踪活细胞中的目标 rna, 感兴趣的 rna 将被标记为 mbs 的多个副本, 以招募荧光蛋白共轭 mcp (mcp-fp), 从而使目标 rna 可见。MS2-MCP系统是最早发明和使用最广泛的RNA追踪系统。之后,建立了各种类似的FE型RNA追踪系统,包括PP7-PCP和λN ( 2 , 3 )。然而,FE 型平台通常具有高背景噪声的弱点。为了提高信噪比 (SNR),使用相当高的拷贝数(通常 >24 拷贝,>1000 bp)的外源串联重复 RBS 来标记目标 RNA ( 1 , 2),这在遗传操作中很复杂,并且可能会增加改变目标 RNA 的结构和定位的可能性。
最近,针对 RNA 成像提出了 Cas9 和 Cas13 基因编辑工具 ( 4-6 )。受益于引导 RNA (gRNA) 介导的 RNA 靶向,FP 缀合的 Cas9/Cas13 可以跟踪任何感兴趣的 RNA。然而,考虑到 MS2-MCP 系统至少需要 12 个 MBS 拷贝才能获得可接受的 SNR ( 1 ),可以想象,Cas9/Cas13 需要针对单个目标 RNA 的不同基序应用相当多的 gRNA RNA 成像平台可实现良好的 SNR。此外,Cas9/Cas13 相对较大的大小是一个问题,因为多个 Cas9/Cas13 报告基因在单个 RNA 上的积累可能会损害目标 RNA 的构象完整性和定位。到目前为止,MS2-MCP 平台仍然是最好的 FE 型 RNA 成像工具。
由于荧光报告基因的应用,所有 FE 型 RNA 跟踪平台都固有地受到高背景噪声的影响。FA 型 RNA 跟踪/成像平台部分解决了这一问题。FA 型平台基于双分子荧光互补 (BiFC)。例如,MCP 和 PCP(一种类似于 MCP 的 RNA 结合蛋白)分别与某个 FP 的 N- 和 C- 片段融合;由此产生的两个报告基因,MCP-FPN 和 PCP-FPC,在它们通过 RNA 靶标上的相邻 MBS 和 PBS(PCP 结合位点)聚集在一起之前,不会重建功能齐全的荧光蛋白(7、8)。因此,FA 型平台的 SNR 大大提高。毫无疑问,当前的 FA 型平台,如 BiFC 或 TriFC RNA 跟踪系统(8),灵敏度超过任何 FE 类型工具。然而,与 MS2-MCP 系统需要的 12 × MBS 标签相比,更大的 RNA 标签,例如 12 × MBS-PBS (1400 nt),经常用于当前的 FA 型平台 ( 7 , 8 )。这种大的外源RNA序列的插入容易引起上述问题。
Cas6 是 IE 型 CRISPR 复合体的核心成分。它通过识别称为 CBS(Cas6 结合位点)的特定茎环 RNA 元件来结合和切割 pre-crRNA ( 9 , 10 )。CBS 结合诱导 Cas6 的变构变化,导致其 N 和 C 末端并列 ( 9 )。利用这一特性,我们将源自大肠杆菌的Cas6 ( Ec Cas6) 蛋白设计为 FA 型报告基因。催化结构域突变的Ec Cas6 (d Ec Cas6) 在其 N 端和 C 端分别附加了蛋白质 Venus、Venus-N (VN) 和 Venus-C (VC) 的非荧光部分。所得嵌合体 VN-d EcCas6-VC 在与感兴趣的 RNA 上的 CBS 结合之前没有荧光。由于荧光互补是由 Cas6 的变构开关介导的,我们将此平台命名为基于 Cas6 的荧光互补 (Cas6FC)。因此,我们设计了一个新的 FA 型 RNA 跟踪平台,具有良好的灵敏度和特异性。
RNA的荧光原位杂交
Hela 细胞和 HEK293T 细胞在 24 孔板中与聚赖氨酸盖玻片一起培养,然后将这些细胞与 200 ng VN-d EC Cas6 -VC-GK 和 800 ng Actin-16 × CBS-GK共转染或 hTERC-16 × CBS-GK。单分子廉价荧光原位杂交 (smiFISH) 用于 RNA 成像 ( 11 )。初级探针和 FLAP 探针的序列列于补充表 S1。根据先前报道的方法进行 smiFISH。简而言之,细胞在 37°C 下用 4% PFA 固定 15 分钟,用 1 × PBS (pH7.4) 洗涤 5 分钟 3 次,然后用 0.5% Triton X-100 透化 15 分钟,这些细胞用 1 × PBS (pH7.4) 重新洗涤 5 分钟 3 次。将细胞在杂交溶液中于 37°C 孵育 12 小时。杂交后,样品用 15% 甲酰胺(在 1 × SSC 中)在 37°C 下洗涤 30 分钟 3 次,在室温下用 0.1 μg/ml DAPI 染色 10 分钟,然后在防褪色封固剂 (Solarbio) 中封固.
流式细胞仪检测
根据实验设计(表1)用质粒转染24孔玻璃底板中的HEK293T细胞。每个实验样品做 4 次重复,其中 2 次用共聚焦显微镜观察,另外 2 次用流式细胞仪分析。在转染后 24 小时,用 0.25% (w/v) 胰蛋白酶(在 1 × PBS pH7.4 中)消化 HEK293T 细胞,并用 BD FACSCelesta 流式细胞仪进行分析。在分析 VN-d Ec Cas6-VC的背景和 CBS 诱导的特异性荧光之前,使用用于红色荧光通道补偿的 Actin-GK + pDsRed-Monomer-C1 转染细胞和 VN-d Ec进行荧光补偿Cas6-VC-GK + Actin-16 × CBS-GK 转染细胞分别用于绿色荧光通道补偿。流式细胞仪分析的参数如下:红色荧光通道 (PE-Texas Red) 的激光电压分别为 440 V 和绿色荧光通道 (FITC) 的激光电压为 320 V。红色荧光通道的荧光补偿参数分别为 4.8 和绿色荧光通道的 12.6。根据FSC(Forward Scatter)和SSC(Side Scatter)从所有细胞中分选出总共10 000个单个活细胞。由于共转染的细胞应表达 DsRed-Monomer 荧光蛋白,因此将红色荧光阳性 (Red + ) 细胞分选并用于分析 VN-d EcCas6-VC 背景和特异性荧光。Actin-GK+pDsRed-Monomer-C1共转染组作为对照组;VN-d Ec Cas6-VC-GK + pDsRed-Monomer-C1共转染组(命名为无CBS组)用于分析初始背景荧光;VN-d Ec Cas6-VC-GK + Rm-20 × CBS-C1组(与CBS组命名)用于分析特异性荧光。Red +对照组细胞的最大荧光强度用作确定初始背景荧光或特异性荧光的阈值。将强度高于上述阈值的红色+无CBS组细胞荧光确定为初始背景荧光;红色+与CBS组细胞的荧光强度高于上述阈值的荧光被确定为特异性荧光。
RNA成像
对于活细胞中的 RNA 成像,转染后 24 小时在蔡司 Vert.A1 显微镜平台上观察 24 孔板中的转染细胞。对于 FISH 成像,将盖玻片安装在蔡司 Vert.A1 显微镜平台上并进行观察。每个荧光通道的曝光时间设置为 5 s,明场设置为 100 ms。共聚焦显微镜观察固定细胞,转染细胞用 4% PFA 37°C 固定 15 min,1 × PBS (pH7.4) 洗涤 5 min 3 次,然后用 0.5 % Triton X-100 15 分钟,这些细胞用 1 × PBS (pH 7.4) 重新洗涤 5 分钟 3 次。用 0.1 μg/ml DAPI 染色并用 1 × PBS (pH7.4) 洗涤 5 min 3 次后,用 Nikon A1R HD25 共聚焦显微镜观察细胞。
结果
大肠杆菌 (Ec) Cas6 可用于体内RNA 追踪
由于其 CBS 结合能力,Cas6 家族的成员有可能用作 FE 型 RNA 跟踪工具。为了测试这种可能性,我们选择了源自大肠杆菌的 Cas6 ( Ec Cas6),并研究了其在生理温度 (37°C) 下追踪哺乳动物细胞中 RNA 的应用。我们构建了一个催化失活的Ec Cas6 突变体(根据之前的研究(12),在第 20 位发生 H→A 突变),并将其命名为 d Ec Cas6(dead Ec Cas6)。确认 a. Ec Cas6 可以识别和切割CBS 和 b. d EcCas6 失去了内切核糖核酸酶活性,但保留了其与哺乳动物细胞中 CBS 的结合活性,我们在从头设计的关闭和打开报告系统中测试它们。在关闭系统中,将 1×CBS 序列插入EGFP mRNA 的 5'-UTR。由于 5' 帽对于 mRNA 翻译至关重要,并且Ec Cas6 介导的切割会从EGFP mRNA中去除 5'-帽,因此成功的Ec Cas6-CBS 相互作用将导致 EGFP 荧光强度降低。在 HEK293T 细胞中,Ec Cas6 表达载体的共转染显着降低了 EGFP 荧光强度,而在使用 d Ec Cas6 表达构建体时未观察到(图1A )。在开启系统中,将16×CBS序列和2×LIN28 RNA核保留信号序列依次插入DsRed-Monomer(简称Rm)mRNA的3'-UTR。LIN28 RNA 核保留信号将宿主 mRNA 保留在细胞核中 ( 13 );因此,无法翻译 DsRed-单体荧光蛋白。通过Ec Cas6 切割去除 LIN28 RNA 核保留信号可以释放DsRed-Monomer mRNA 以在细胞质中进行翻译。事实上,当通过荧光显微镜评估时,Rm-CBS-LIN28-C1 载体的转染显示出几乎无法检测到的 DsRed-单体蛋白表达。然而,引入EcCas6 显着增强了 DsRed-Monomer 蛋白的荧光,反映了成功的Ec Cas6 介导的切割。正如预测的那样,将 d Ec Cas6 引入细胞不能开启 DsRed-单体蛋白的表达(图1B)。总之,我们验证了Ec Cas6-CBS 系统在哺乳动物细胞中的有效性,并且 d Ec Cas6 确实失去了酶活性。

大肠杆菌Cas6 ( Ec Cas6) 可用于真核 RNA 成像。( A , B )使用 (A) '关闭' CBS-EGFP报告系统或 (B) '开启' DsRed-单体-CBS 验证Ec Cas6 和 d Ec Cas6的 CBS 结合活性和催化活性HEK293T 细胞中的报告系统。将不包含 LIN28 或 CBS 序列的单独的 RFP/EGFP 表达载体作为内部对照进行共转染。比例尺,200 μm。( C ) HEK293T 细胞与表达DsRed-单体的载体共转染对附加 (Rm-20 × CBS-C1) 或不附加 (pDsRed-Monomer-C1) CBS 序列的 mRNA 和表达Ec Cas6-EGFP 或 d Ec Cas6-EGFP 的质粒进行成像,以了解DsRed-Monomer mRNA 细胞内分布。低功率图像的比例尺,50 μm;高倍图像的比例尺,10 μm。所用质粒的剂量列于表1。来自 3 次独立实验的代表性图片。d Ec Cas6 上的突变只会消除其核糖核酸内切酶活性,并保持 CBS 结合能力。利用 d Ec Cas6的这一特性,我们用 EGFP 对 d Ec Cas6 进行基因标记,并使用这种荧光嵌合蛋白来跟踪用 CBS 进行基因标记的目标 RNA。如图1C所示,在目标DsRed-Monomer mRNA上没有 20 × CBS 标签时, Ec Cas6-EGFP 和 d Ec Cas6-EGFP 在细胞质和细胞核中都可以看到。将 20 × CBS 标签挂在目标 mRNA 上,只有用 d Ec Cas6-EGFP 共转染的细胞,而不是用EcCas6-EGFP 构建体可以将目标 mRNA 定位在细胞质中。因此,d Ec Cas6,当用荧光蛋白标记时,可作为 FE 类型工具用于 RNA 检测。
基于VN-d Ec Cas6-VC的FA型RNA追踪平台的建立
与其他 FE 型 RNA 跟踪平台类似,d Ec Cas6-EGFP 技术也存在信噪比不佳的高背景问题。据报道,与 CBS 的结合诱导了嗜热栖热菌Cas6 ( Tt Cas6) 的构象变化,导致 Tt Cas6 的 N 和 C 末端并列( 9 ) (补充图S19)。这一发现促使我们研究 Cas6 可被用作体内追踪 RNA 的 FA 平台的可能性。然而, Tt Cas6 的最佳工作温度约为 65°C,无法在哺乳动物细胞中使用。大肠杆菌在 37°C 下繁殖。基于Ec Cas6 和Tt Cas6 之间结构的相似性 (补充图 S19B)( 14),我们假设Ec Cas6 可以在哺乳动物细胞中用于此目的。
为了构建 FA 型荧光报告基因,我们将分裂的 FP 片段与 d Ec Cas6 蛋白的末端用不同的肽接头连接,产生一系列 d Ec Cas6 分裂的 FP 融合蛋白。补充图 S20-S22)。FA 型荧光报告基因在游离状态下应该是非荧光的,但在结合目标 RNA 时会变成荧光。根据该标准,分离出一种所需的融合蛋白,称为 VN-d Ec Cas6-VC (53 kDa)。
这种 VN-d Ec Cas6-VC 融合蛋白由顺序连接的 Venus-N 末端片段 (1-153)、linker1、d Ec Cas6、linker2 和 Venus-C 片段 (154-238) 组成(图2A和补充图S7)。为了证明概念验证,我们用表达 VN-d Ec Cas6-VC 的质粒转染 HEK293T 细胞;一起共转染表达天然DsRed-单体mRNA 或 CBS 标记的 DsRed-单体mRNA 的载体。20×CBS 标签附加在 mRNA 的 3' 末端,以尽量减少其对 RNA 构象和分布的影响。当转录未标记的 DsRed-Monomer mRNAVN-d Ec Cas6-VC 不显示自发荧光(图2B)。然而,当 CBS 标记的DsRed-Monomer可以在细胞质中成功检测到荧光金星的信号使用了mRNA。在 3' 末端添加 20 × CBS 标签不会影响 mRNA 的翻译,因为在两组之间可以观察到相当的 DsRed-单体蛋白信号(图2B)。当测试 COS-7 和 Hela 细胞系时,获得了类似的结果(补充图 S23),表明基于 d Ec Cas6 的荧光互补 RNA 跟踪平台对广泛的哺乳动物细胞的适用性。不同于以前的 BiFC 或 TriFC FA 型 RNA 跟踪平台,其荧光互补是通过与其 RBS 并列结合的间接蛋白质-蛋白质相互作用介导的;VN-d Ec Cas6-VC 是一种单蛋白、RBS 触发、基于变构开关的 FC(荧光互补)RNA 跟踪平台。由于 Cas6 蛋白赋予其所有独特特征,我们将这个新的 RNA 跟踪平台命名为基于 Cas6 的荧光互补 (Cas6FC)。
图 2。

VN-d Ec Cas6-VC 可用于 FA 模式 RNA 成像。( A ) Cas6FC 平台的 RNA 追踪推测机制图解。( B )在 HEK293T 细胞中用 Cas6FC可视化DsRed-Monomer-20 × CBS mRNA。低功率图像的比例尺,50 μm;高倍图像的比例尺,10 μm。( C ) FISH 验证 Cas6FC 介导的 RNA 跟踪策略的图解说明。( D )来自 Cas6FC 和 FISH的ACTB mRNA 跟踪信号在 HEK293T 细胞中的相关性。比例尺,20 μm。( E ) hTERC的相关性lncRNA 跟踪来自 HeLa 细胞中 Cas6FC 和 FISH 的信号。比例尺,20 μm。所用质粒的剂量列于表1。来自 3 次独立实验的代表性图片。最后,为了验证 Cas6FC RNA 追踪的真实性,我们对 Cas6FC 平台和 RNA smiFISH(单分子廉价荧光原位杂交)技术进行了并排比较。为了排除它们之间的技术差异带来的潜在偏差,FISH探针针对目标RNA的天然非CBS序列设计,同一批细胞同时进行了Cas6FC和RNA smiFISH测试(图2C)。在这种情况下,FISH 探针与 Cy5(一种发射波长远离金星的荧光团)连接,从而实现共定位分析。HEK293T细胞的ACTB mRNA和hTERC(人端粒酶RNA成分)选择Hela细胞的lncRNA作为比较的靶RNA。ACTB的 mRNA定位于细胞质并编码管家细胞骨架蛋白 ( 15 ),而定位于细胞核的hTERC RNA 与癌细胞增殖有关 ( 16 )。通过共聚焦显微镜检查两种方法的标记模式。Cas6FC 和 RNA smiFISH 在跟踪两个目标的 RNA 时高度一致的渲染(图2D和 E) 保证使用 Cas6FC 平台探测哺乳动物细胞中的 RNA。此外,这些结果表明,在 3' 末端标记 CBS 的策略不会影响靶向 RNA 的自然定位。
Cas6FC 是一种灵敏的 RNA 追踪系统
为了进一步评估 Cas6FC 平台在跟踪目标 RNA 方面的灵敏度,我们使用了流式细胞仪,它在区分信号和噪声方面比荧光显微镜更敏感。与肌动蛋白-GK 载体(表达 β-肌动蛋白作为对照)和 pDsRed-单体-C1 载体(表达DsRed-单体mRNA 和蛋白)共转染的细胞提供的背景参考相比,与VN-d Ec Cas6-VC-GK 载体(表达 Cas6FC 平台的报告基因)和 pDsRed-Monomer-C1 载体几乎没有可检测到的金星信号(0.78% 的阳性细胞与 0% 的背景)(图3A)。相比之下,与 VN-d Ec共转染的组中有 41.9% 的细胞Cas6-VC-GK 载体和 Rm-20 × CBS-C1 载体(表达DsRed-Monomer-20 × CBS mRNA 和 DsRed-Monomer 蛋白)显示出强烈的金星荧光。因此,在目标 RNA 中没有 CBS 序列的情况下,VN-d Ec Cas6-VC 产生的噪音可以忽略不计。
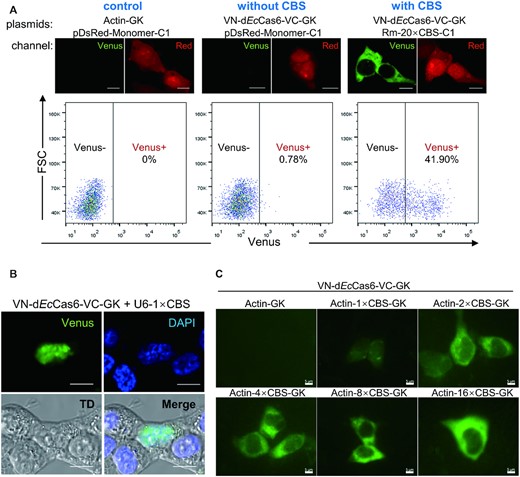
Cas6FC 平台的灵敏度。(一)用表达pDsRed-Monomer-C1或Rm-20×CBS-C1的载体转染HEK293T细胞。还用表达 β-肌动蛋白(作为对照)或 VN-d Ec Cas6-VC 的质粒转染细胞。24 小时后,通过共聚焦显微镜(上图)分析 DsRed 单体和金星的表达。平行地,红细胞被门控以通过流式细胞术分析金星信号。显示了来自 3 个独立实验的代表性图片。( B ) 在 HEK293T 细胞中,在 RNA 聚合酶 III 启动子下转录了一个仅携带一个 CBS 拷贝的 29nt 长的 RNA。Cas6FC 系统可以将其定位在细胞核中。比例尺,10 μm。(三)ACTB携带 0×、1×、2×、4×、8× 或 16× CBS 的 mRNA 在 HEK293T 细胞中的 RNA 聚合酶 II 启动子下转录。通过荧光显微镜检查 Cas6FC 系统对 CBS 数的敏感性。比例尺,5 μm。所用质粒的剂量列于表1。显示了来自三个独立实验的代表性图片。简单地增加 RBP 结合基序的拷贝数可以提高 FE 型 RNA 跟踪平台的灵敏度。例如,MS2-MCP 系统中经常使用 24 × MBS(MCP 结合位点)来获得最佳信号(1)。然而,将多个 RBS 拷贝插入目标 RNA 会改变 RNA 的结构和/或分布的担忧始终困扰着。因此,我们准备确定 Cas6FC 平台所需的最小 RBS(在本例中为 CBS)副本。为此,我们首先测试了 Cas6FC 检测仅携带一个 CBS 拷贝的 29nt 长 RNA 的敏感性。该短 RNA 在人 U6 小核 RNA 启动子(RNA 聚合酶 III 启动子)下转录,该启动子在生成具有高输出的短转录本方面具有优势。令我们惊讶的是,在 HEK293T 细胞中可以明显看到 1×CBS RNA(图3B)。该信号仅位于细胞核,这与 U6 启动子驱动的 RNA 缺乏 mRNA 的 5' 帽和 Poly A 尾并优先分布在细胞核中的事实一致。接下来,我们检查了 CBS 对检测由 CMV 启动子(RNA 聚合酶 II 启动子)驱动的靶 mRNA 的剂量效应(图3C)。荧光强度以 CBS 拷贝数依赖性方式增加,并且在 Cas6FC 平台中再次可见携带一个 CBS 拷贝的 mRNA。总之,小至 1 × CBS (∼29nt) 就可以赋予 Cas6FC 平台的可检测性。插入这样一个短标签可能会简化基因操作,同时显着降低干扰目标 RNA 折叠和分布的可能性。(后续内容见原文------)
原文:https: //doi.org/10.1093/nar/gkac014